The research team of JI Weikai from the School of Basic Medical Sciences/National Key Laboratory for the Diagnosis and Treatment of Zoonotic Infectious Diseases at Huazhong University of Science and Technology published their paper entitled "Sec23IP recruits VPS13B/COH1 to ER exit site-Golgi interface for tubular ERGIC formation" in the Journal of Cell Biology. In their research, the team identified Sec23IP, an ER exit site-associated protein, to be a recruitment factor for VPS13B. Sec23IP recruits VPS13B to the ER exit site-Golgi interaction interface. While prior studies have established that VPS13B and Sec23IP played a pivotal role in sperm acrosomal biogenesis, their physical interaction has not been clarified. Their study revealed the key role of VPS13B in the early secretory phase from the ER to the Golgi, and suggested that early secretory defects are a common factor leading to the pathogenesis of Cohen syndrome and abnormal sperm acrosomal biogenesis.

The life process of eukaryotic cells depends on specialized organelles. Different organelles have discrete functions but also have to work together to form an organelle interaction network. Organelles engage in rapid material exchange and information communication through physical contact (membrane contact), efficiently performing various biological functions and functionally maintaining cellular and organismal homeostasis. Transmembrane proteins play a key part at these membrane contact sites. Transmembrane proteins specifically enrich at membrane contacts by mediating membrane lipid exchange between organelles, regulating the biogenesis, degradation, and dynamics of organelles.
VPS13B is a member of the transmembrane protein family. The human genome contains four VPS13 genes (VPS13A-D), which are directly related to various diseases, making these proteins of significant research interest and a hot topic in the academic communities. Studies have shown that VPS13A, VPS13C, and VPS13D are transmembrane proteins of the ER-associated membrane contact sites (MCS), including ER-mitochondria/plasma membrane (VPS13A) and ER-late endosome/lysosome MCS (VPS13C). VPS13D plays a role in Drosophila mitophagy and is localized at ER-mitochondria/peroxisome contacts. During starvation, VPS13D collaborates with the ESCRT protein TSG101 to regulate lipid droplet remodeling.
Cohen syndrome represents a rare autosomal recessive developmental disorder with diverse clinical manifestations, primarily including developmental and mental retardation, joint hypermobility, microcephaly, facial dysmorphism, progressive pigmentary retinopathy, severe myopia, and intermittent neutropenia.
Loss-of-function mutations in the VPS13B gene are the only known cause of Cohen syndrome, but the cellular mechanisms of its pathogenesis are not well understood. Although VPS13B has been shown to play an important role in multiple cellular processes, such as Golgi integrity and neurite growth, cargo retrieval/transport, acrosomal biogenesis, and lipid droplet dynamics, and compared to other VPS13 proteins, the current the study regarding VPS13B remains at the stage of functional description. At present, the correlation between VPS13B and MCS and the pathogenesis of VPS13B-related loss-of-function genetic diseases are subjects of active scientific investigations.
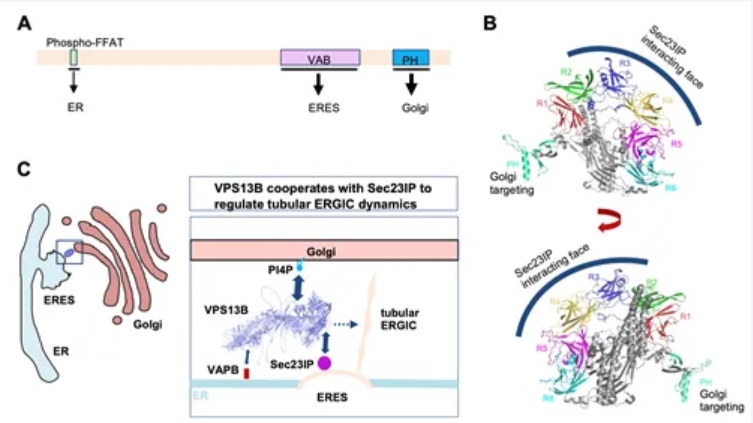
The research team delved into the molecular mechanisms of the interaction between Sec23IP and VPS13B, and found that the VAB (VPS13 adaptor binding) domain of VPS13B was responsible for binding to the N-terminal disordered region of Sec23IP. This discovery is consistent with the well-accepted mode of interaction between VPS13 proteins and their recruitment factors. Importantly, researchers clarified, through imaging and biochemical experiments, that Cohen syndrome-related VPS13B mutations significantly inhibited the interaction between VPS13B and Sec23IP, suggesting that alternations in their interaction may be a potential pathogenic factor.
VPS13 proteins can bridge two organelles at membrane contact sites, and transport lipids, thereby enabling the growth and expansion of organelle membranes and promoting organelle biogenesis. Their study found that the absence of VPS13B did not significantly affect the morphology, abundance, and distribution of ERES, coatomer, and classical ERGIC, but significantly inhibited the formation of a new type of cargo transport carrier, the tubular ERGIC. Complementation experiments showed that VPS13B promoted the occurrence of this new type of cargo transport carrier in a manner dependent on its transmembrane activity. Furthermore, primary culture of mouse MEF cells, imaging and omic analyses exhibited that VPS13B and Sec23IP participated in promoting the transport of collagen precursors from the ER to the Golgi, suggesting that the hypermobility of joints in Cohen syndrome patients may be related to abnormal collagen secretion.
This study identified Sec23IP as a recruitment factor for VPS13B, elucidated the localization and molecular mechanisms of VPS13B, and provided clues to an issue of long-standing concern in the field. It revealed that VPS13B acted as a regulator of ER-to-Golgi transport, meeting the high-intensity transport and secretion needs of specific tissues and cells (such as nerves, joints, and sperm, etc.) during organism development. Researchers believe that cargo transport abnormalities caused by VPS13B genetic mutations (including but not limited to collagen) may be a mechanism underlying development of Cohen syndrome. Although this study suggests that the transmembrane activity of VPS13B is crucial for the occurrence of tubular ERGIC, the link between the two has yet to be found, and the mechanism of VPS13B lipid transport remains to be clarified. In summary, this study provides a new perspective and basis for exploring the mechanisms of rare diseases being associated with mutations in the VPS13 family of transmembrane proteins.
DU Yuanjiao and FAN Xinyu, two doctoral students at the School of Basic Medical Sciences of Huazhong University of Science and Technology, are the first authors of the paper, and Professor JI Weikai is the corresponding author. Researcher DENG Lin from Shenzhen Bay Laboratory provided substantial assistance in the implementation of this project. This study was funded by the National Natural Science Foundation of China and the Shenzhen Bay Tongzhou Scholar Program.
JI Weikai's team has long been engaged in the study of organelle interactions and the pathogenesis of related diseases, their work revolving around "membrane contact and lipid transport," achieving a series of innovative results, identifying key new factors in the occurrence and regulation of organelle membrane contacts, and revealing the cellular biological mechanisms of diseases caused by mutations in these key factors, providing a theoretical foundation for looking into the shared mechanisms of transmembrane protein mutations.The laboratory is currently looking for postdoctoral fellows in the fields of cell biology, biochemistry, immunology, and proteomics/lipidomics to join their team.








 Chinese
Chinese
